服务热线:
18952377777

联系人:徐煦
电话:18952377777
固话:0517-85222222
联系人:陆朗辉(长江以北地区)
电话:17625179812
固话:0517-85322222
联系人:梁言(长江以南地区)
电话:18005236279
固话:0517-85200000
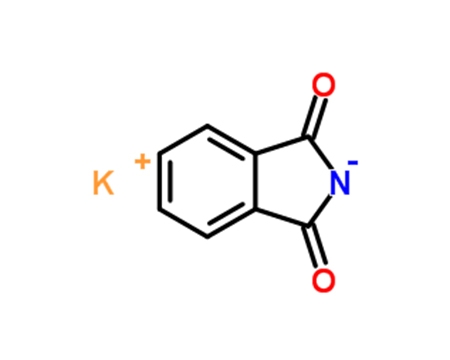
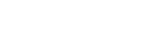
3-(2-硝基苯氧基)-1-丙胺的合成是由2-硝基苯酚钠在无水DMF中,KI为催化剂与N-(3-溴丙基)邻苯二甲酰亚胺反应,合成2-[3-(2-硝基苯氧基)-丙基]二氢异吲哚-1,3-二酮(4a),再经肼解制得[10]。
由于N-(3-溴丙基)邻苯二甲酰亚胺和生成物中的邻苯二甲酰亚胺都能在碱性条件下水解,所以反应不能在水中进行,要用无水溶剂。该方法操作要求较高,试剂较贵。我们以取代的2-硝基苯酚为原料,NaOH为碱,与1,3-二溴丙烷反应制备3-芳氧基-1-溴丙烷,再经Gabriel反应合成3-芳氧基-1-丙胺,并对反应条件进行了研究。取代的2-硝基苯酚与1,3-二溴丙烷的反应可以在常规加热和微波辐射加热下进行[13]。
结果显示,该步反应在常规加热下需要5h完成,在微波辐射加热下只需10min就可结束,反应速度明显加快,收率有所提高。按文献用常规加热方法合成时,反应液为两相,通过剧烈搅拌和加入PTC使反应加速,但1,3-二溴丙烷和3-芳氧基-1-溴丙烷的水解也加快,影响反应收率。通过摸索研究发现,先在少量水中制成取代的2-硝基苯酚钠,再加入1,3-二溴丙烷和四丁基溴化铵,经超声分散均匀后,置于微波反应器中,功率130W,辐射10min反应即可结束,收率较高。
功率为65W时反应速度较慢,功率为300W时有聚合物生成。由于水量少,反应基本上是在无溶剂下进行的,这样既可以使反应加快,又可以降低原料和生成物的水解,使收率增加。1,3-二溴丙烷具有两个取代反应点,能够与取代的硝基苯酚钠形成双取代的产物,所以在反应时加大1,3-二溴丙烷的投料量,以减少双取代产物的生成,当取代的硝基苯酚与1,3-二溴丙烷的摩尔投料比为1∶2时,常规加热和微波辐射加热反应时只有少量的双取代产物生成。

化合物5a~5f的合成中,肼解生成的白色絮状沉淀不溶于氯仿,反应物经过滤,减压蒸出乙醇后,加氯仿溶解,再过滤,水洗涤即可得到3-芳氧基-1-丙胺。成盐酸盐时可直接在氯仿液中加适量浓盐酸摇匀,减压蒸出氯仿,残留物加丙酮分散得到相应的盐酸盐。
化合物4a~4f在测定HRMS(EI)时未能得到分子离子峰,4a~4f都给出了m/z180,160碎片离子峰,这与化合物结构在邻苯二甲酰亚胺片段中氮原子的β位及芳醚氧原子的α位容易发生裂解,形成包含邻苯二甲酰亚胺片段的碎片离子峰一致。4a~4f元素分析结果与化合物的结构完全吻合。所有化合物的结构均经FTIR,1HNMR,13CNMR,HRMS或元素分析确证。
目标化合物的生物活性及影响因素的讨论
通过ELISA法测定不同浓度的目标化合物和biotinylatedTat-AcK50组氨酸竞争性地与固定在96孔板中的GST-fusionBRD结合,来确定目标化合物的体外抑制活性。测定结果见表1。体外抑制活性测定结果显示,化合物5a~5f在较高浓度下,能够抑制HIV-1AcTat的活性。但与N1-(2-硝基芳基)-1,3-丙二胺类盐酸盐的体外抑制活性相比,有了明显的下降。
在详细分析N1-(4-甲基-2-硝基苯基)-1,3-丙二胺盐酸盐(1b)与PCAFBRD复合物3D结构的基础上,认为该化合物的抑制活性主要来源于2-硝基与Y802和Y809的酚羟基相成氢键(d1=2。85Å,d2=3。09Å),以及1,3-丙二胺支链末端铵盐离子与E750的羧基离子形成的盐桥作用(d=2。92Å)(图2A)。2位硝基处于与苯环交叉位置,有利于与靶点中蛋白质氨基酸残基Y802和Y809的酚羟基形成氢键。
为了分析化合物5a~5f生物活性下降的原因,我们用软件对化合物1b和5b的优势构象进行计算,结果显示1,3-丙二胺支链末端铵盐离子均与2位硝基形成氢键。但化合物5b中氢键作用(d=1。88Å)(图2B)要强于化合物1b(d=2。25Å)(图2C)。

当化合物分子进入蛋白靶点后与蛋白质中氨基酸残基发生相互作用时,首先要破坏1b和5b中1,3-丙二胺支链末端铵盐离子与2位硝基形成的氢键。由于化合物5b中氢键作用很强,难以断开,因而影响了该化合物与靶点中蛋白质氨基酸残基的相互作用。
同时该氢键作用还影响到2位硝基的空间取向,使其几乎处于与苯环共平面状态,难以同时与靶点中蛋白质氨基酸残基Y802和Y809的酚羟基形成氢键作用。这些因素可能是造成3-芳氧基-1-丙胺类盐酸盐活性下降的主要原因。
研究表明,N1-(2-硝基芳基)-1,3-丙二胺类盐酸盐作为HIV-1Tat/PCAFBRD抑制剂具有高度的结构专一性,改变为3-芳氧基-1-丙胺类盐酸盐时,活性有所下降。该研究对HIV-1Tat/PCAFBRD抑制剂的进一步优化设计具有一定的指导意义。